Our Services
VANGARD provides standard analysis for RNA-Seq, DNA-Seq, ChIP-Seq, and GRO/PRO-Seq, which implements "best practices" pipeline for the sequencing data provided.
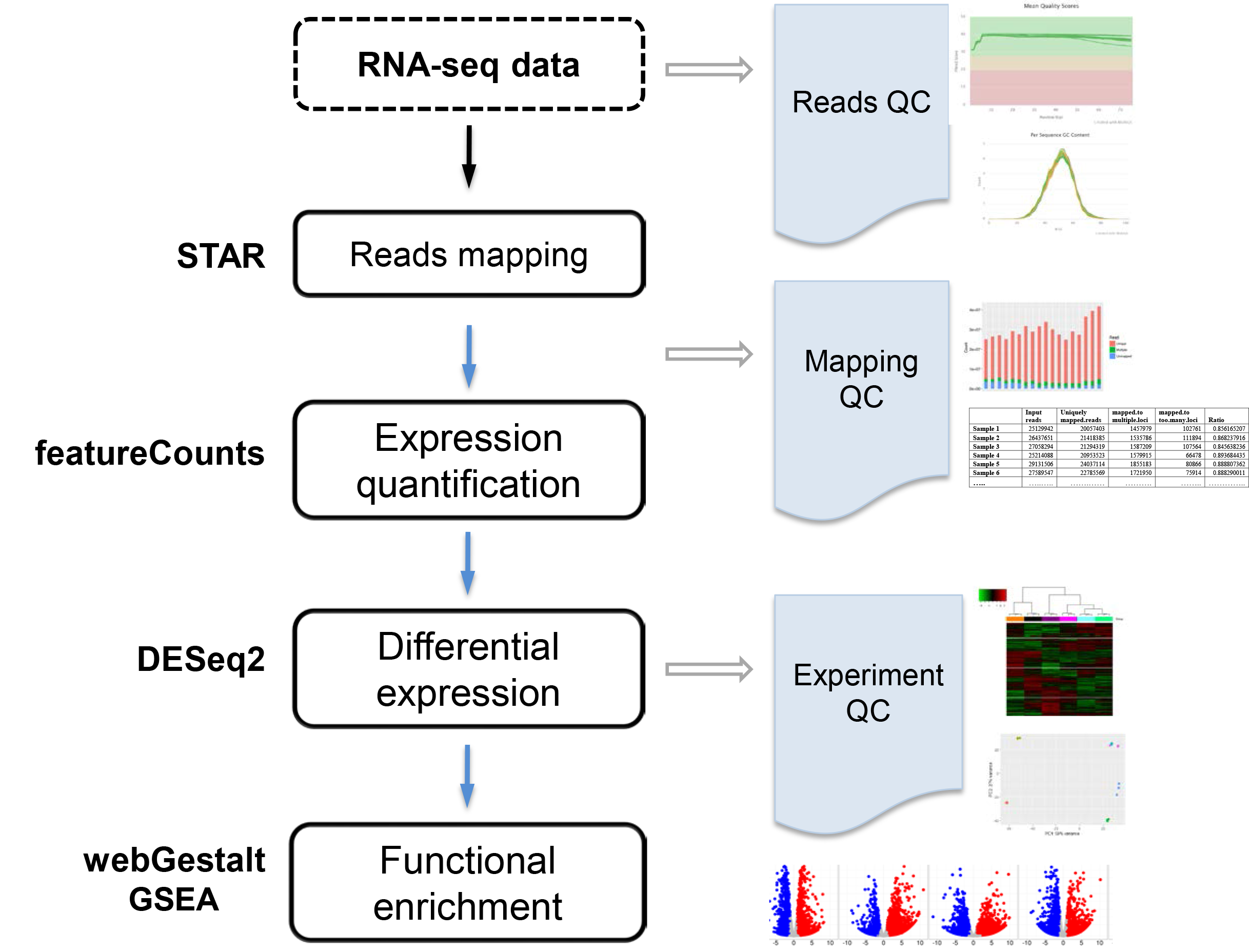
RNA-Seq Analysis
The goal is to identify differentially expressed genes across conditions. RNA-seq data are mapped to the reference genome using STAR, gene expression are quantified by featureCounts, differentially expressed genes are identified by DESeq2, and enriched function or pathways are discovered by webGestalt. VANGARD will perform quality check, including raw reads quality (reads QC), mapping quality (mapping QC) and experiment quality (experiment QC) to ensure high quality results.
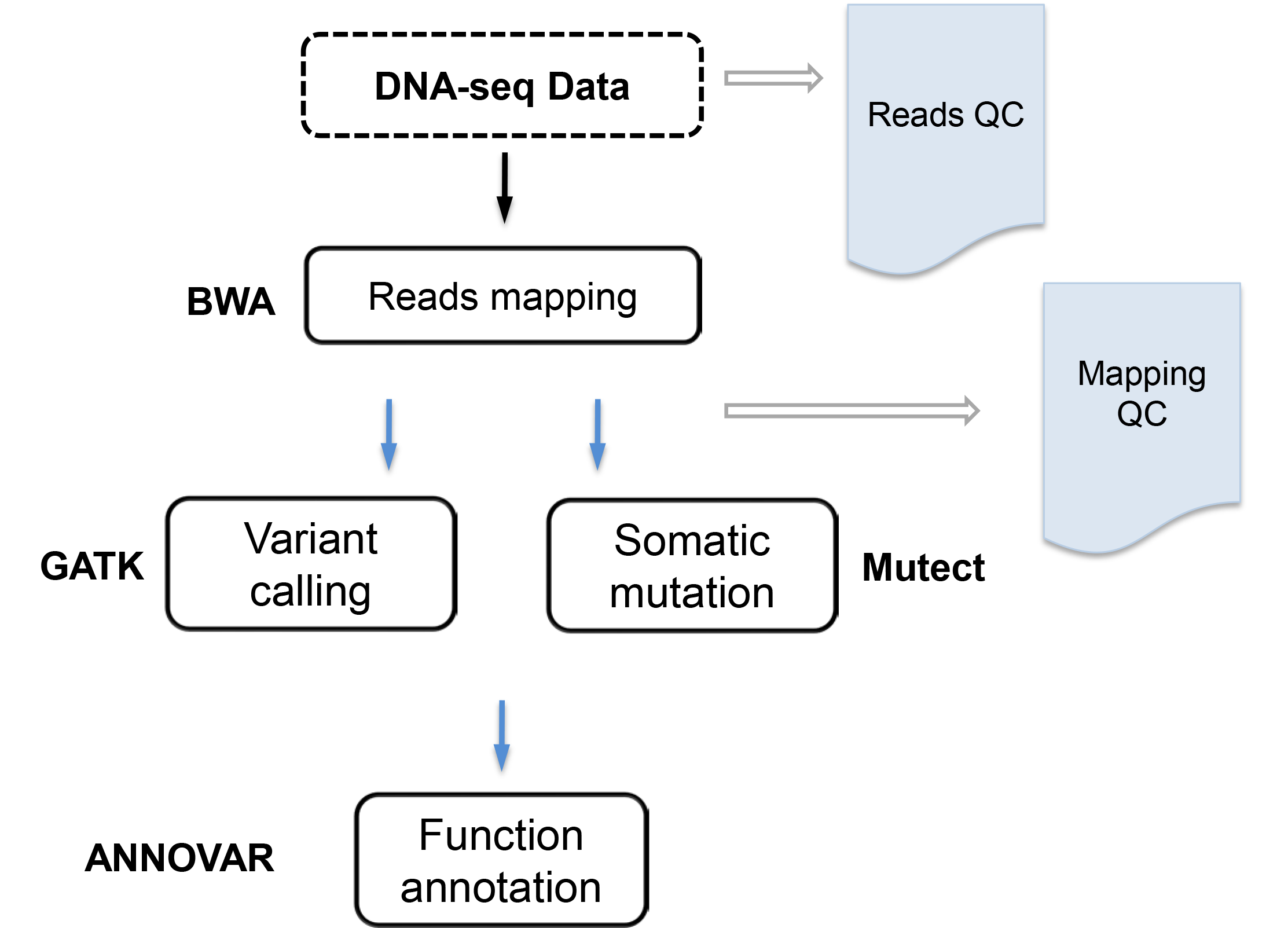
DNA-Seq Analysis
The goal is to call variants or somatic mutations. DNA-seq data are mapped to the reference genome using BWA, variants are called using GATK Best practice, somatic mutation are identified by Mutect, and variants/somatic mutations are functionally annotated by ANNOVAR. VANGARD will perform quality check, including raw reads quality (reads QC), mapping quality (mapping QC) to ensure high quality results.
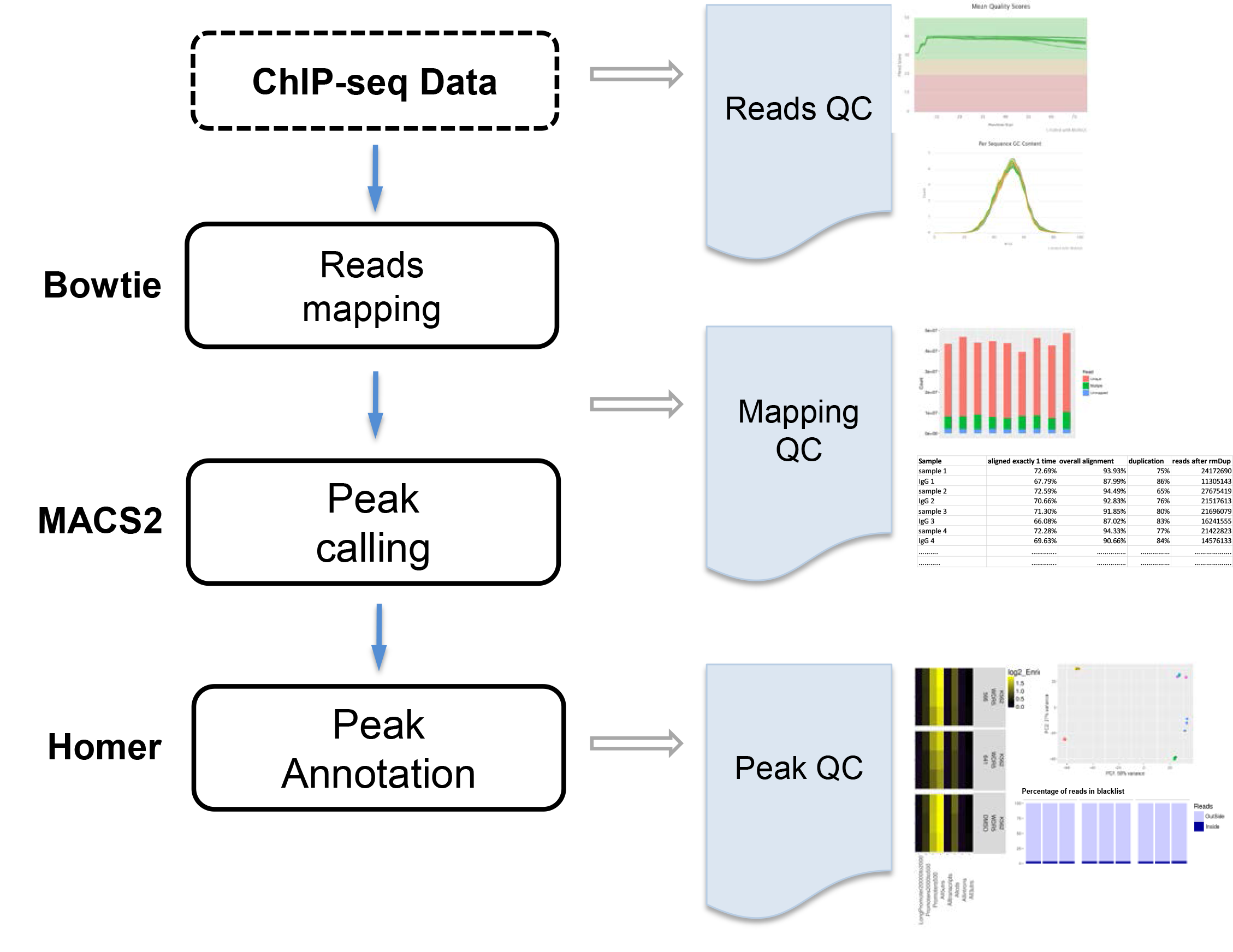
ChIP-Seq Analysis
The goal is identify protein binding sites. ChIP-seq data are mapped to the reference genome using Bowtie, peaks are called using MACS2, and peaks are annotated by Homer. VANGARD will perform quality check , including raw reads quality (reads QC), mapping quality (mapping QC) and peak quality (peak QC) to ensure high quality results.
GRO-/PRO-Seq Analysis
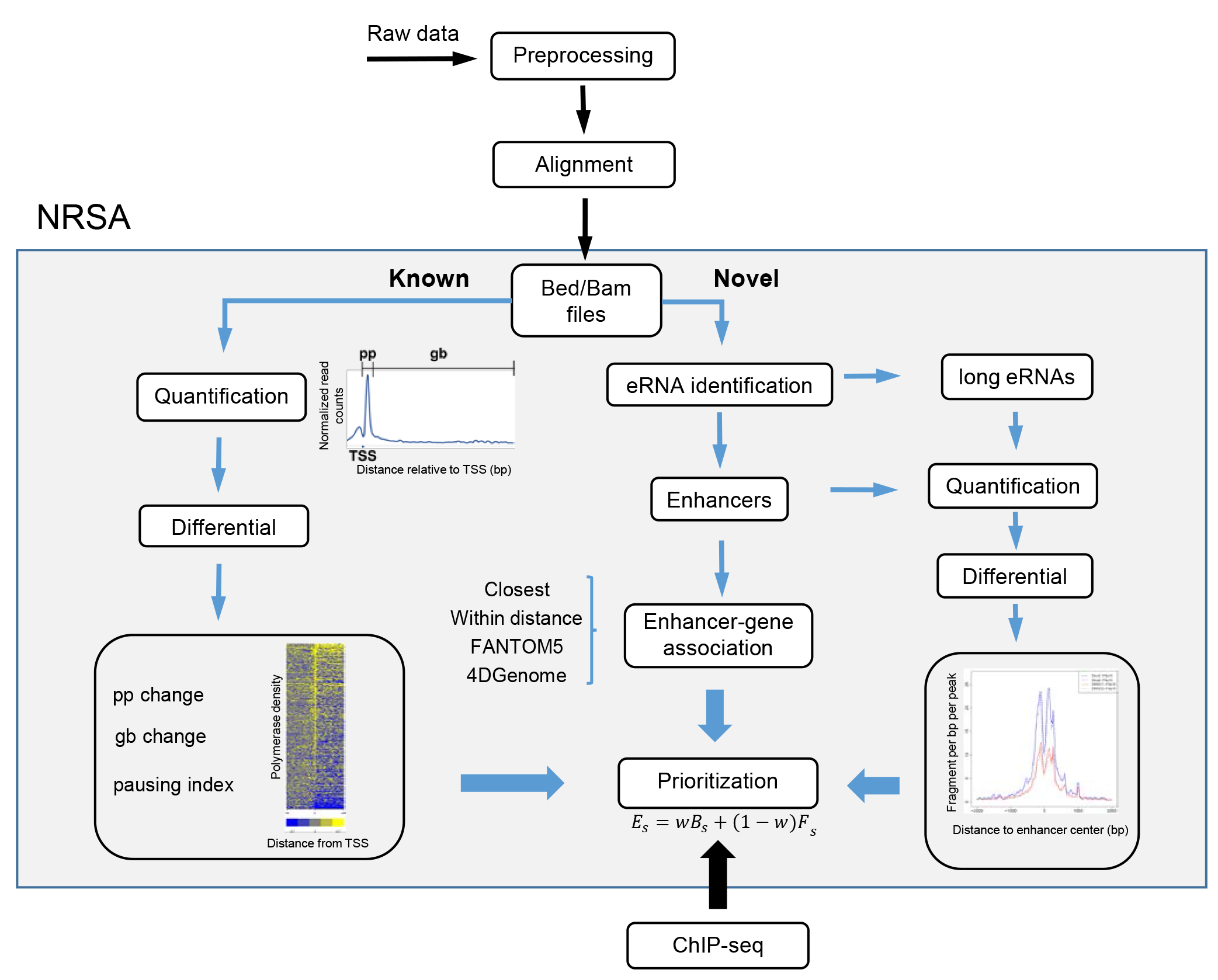
The goal is to identify nascent transcription changes for genes and enhancers. We use a toolkit (named NRSA) developed by ourselvies to do PRO-/GRO-seq data analysis.