Introduction
eNRSA is an enhanced version of NRSA for analyzing nascent transcription profiles generated by PRO-seq, GRO-seq, (m)NET-seq, and Butt-seq data.
The source code of eNRSA is available at GitHub.
The Docker image is available at DockerHub
eNRSA is licensed under the GNU General Public License v3.0
Citation
Highlights
Achieves substantial performance enhancements (~20x faster with markedly reduced memory usage).
Compatible with any species using GTF and FASTA reference files.
Supports multi-factor experimental designs, with adjustments for confounding factors.
Enables identification of alternative transcription start sites (TSS) and termination sites (TTS).
Provides analysis of readthrough transcription dysregulation.
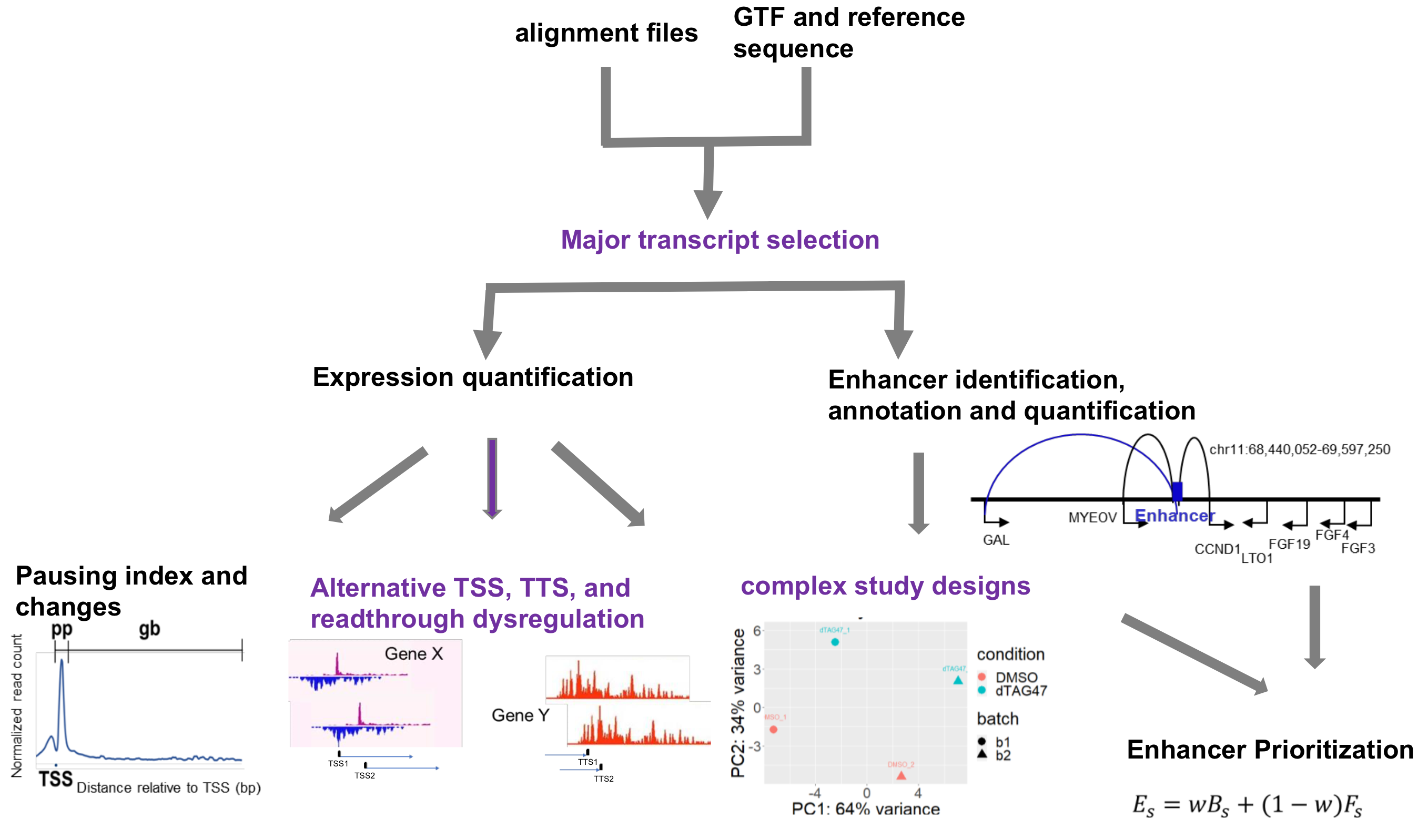
There are two ways for using eNRSA:
Command line usage – download eNRSA code and install the dependencies (see Installation and Usage).
Docker or Singularity container (see Container Usage).
Installation
Local Installation
eNRSA has the following dependencies:
Dependencies
Python packages:
fisherpandasnumpypydeseq2matplotlibscipystatsmodels
Other Packages:
bedtoolsHOMER
Fisher package
The version of fisher available on PyPI is outdated and migth fail to install when using the command pip install fisher.
Please use either of these two solutions to resolve this:
Download the package from its GitHub repository and install it locally:
git clone https://github.com/tylerjereddy/fishers_exact_test.git pip install ./fishers_exact_test
Use
condato install it:conda install -c conda-forge fisher
Both methods ensure you have a functional and up-to-date version of the fisher package.
We recommend using conda to install the dependencies. Follow these steps:
# Create an environment for eNRSA
conda create -n enrsa python=3.12 -y
conda activate enrsa
# Install dependencies
conda install -y fisher pandas numpy pydeseq2 matplotlib scipy statsmodels bedtools homer
eNRSA initialization
# Clone the eNRSA repository
git clone https://github.com/chc-code/eNRSA.git
# Download and unzip reference files
cd eNRSA
wget https://bioinfo.vanderbilt.edu/eNRSA/download/eNRSA_ref.zip
unzip eNRSA_ref.zip
rm eNRSA_ref.zip
Docker / Singularity Installation
You can use pre-built Docker or Singularity images, which includes all dependencies and reference files.
# Docker
docker pull chccode/enrsa:latest
# Singularity
singularity build enrsa.sif docker://chccode/enrsa:latest
Input Files
Alignment Files
Use the -in1 and -in2 options to specify alignment files for control and case samples, respectively. For multiple files, separate them with spaces.
Supported file formats:
BAM
BED
Design Table
For complex experimental designs with cofounding factors, it’s better to define design table using -design <file_path>. This option overrides -in1 and -in2.
Format
The design table is a tab-delimited text file with two sections:
Sample Information:
Column 1: Full path to the alignment file (required)
Column 2: Group name, used to define comparisons (required)
Column 3: cofounding factor (optional)
Comparison Definitions:
Each row starts with
@@.The first column specifies the case group name.
The second column specifies the control group name.
Example:
/nobackup/INF_Cre_0hr_1-R1.bam INF_Cre_0hr b1
/nobackup/INF_Cre_0hr_2-R1.bam INF_Cre_0hr b2
/nobackup/INF_EV_0hr_1-R1.bam INF_EV_0hr b1
/nobackup/INF_EV_0hr_2-R1.bam INF_EV_0hr b2
@@INF_Cre_0hr INF_EV_0hr
GTF File
eNRSA has 8 built-in organisms (hg19, hg38, mm10, mm39, dm3, dm6, ce10, danrer10). For other organisms or custom annotations, useres need to specify a GTF file using -gtf <file_path>.
Format
Rows with
exonas the third column (feature) will be processed.The ninth column (attributes) must include
transcript_idandgene_name.
Output
eNRSA generates a variety of results, including:
Gene-Level Metrics: Nascent RNA abundance in promoter-proximal and gene body regions, pausing index and pausing significance.
Enhancer and eRNA Detection: Identified active enhancers, long eRNAs, and their quantification.
Differential Analysis: Transcriptional changes in promoter-proximal, gene body, and enhancer regions, and pausing index changes across conditions.
Visualizations: Plots offering a global view of the data.
Results for known genes are stored in the known_gene folder, and results for enhancers are stored in the eRNA folder.
known_gene folder
| File Name | File Description |
|---|---|
| pindex.txt | Pausing information for each gene in all samples |
| normalized_pp_gb.txt | Normalized read counts in promoter-proximal and gene body regions for each gene in all samples |
| pp_change.txt | Differential expression results of genes within promoter-proximal region across two conditions |
| gb_change.txt | Differential expression results of genes within gene body region across two conditions |
| pindex_change.txt | Differential expression results of genes of pausing index across two conditions |
| boxplot_ppdensity.pdf | Box plot of normalized read density of promoter-proximal regions for each sample |
| boxplot_gbdensity.pdf | Box plot of normalized read density of gene body regions for each sample |
| boxplot_pausingIndex.pdf | Box plot of pausing index for each sample |
| pindex_change.pdf | Heatmap of pausing index change across two conditions for genes with adjp < 0.05 |
| heatmap.pdf | Heatmap of condition-dependent transcription changes around TSS for active genes |
| Reps_condition1.tif | Histogram for variation across samples within condition 1 |
| Reps_condition2.tif | Histogram for variation across samples within condition 2 |
| TSS_alternative_isoforms_between_conditions.sig.tsv | Differential TSS usage across two conditions |
| TTS_alternative_isoforms_between_conditions.sig.tsv | Differential TTS usage across two conditions |
| readthrough_change.txt | Dysregulated readthrough between two conditions |
eRNA folder
| File Name | File Description |
|---|---|
| Enhancer.txt | List of identified enhancers with annotation, predicted target genes from different strategies, and rank scores |
| Enhancer_center.txt | List of enhancer centers |
| normalized_count_enhancer.txt | Normalized counts for each enhancer |
| Enhancer_change.txt | Differential expression results of enhancers across two conditions |
| long_eRNA.txt | Identified long eRNAs (default: length > 10 Kb) |
| longeRNA-pindex.txt | Pausing information of long eRNAs for all samples |
| longeRNA-normalized_pp_gb.txt | Normalized read counts in promoter-proximal and gene body regions of long eRNAs |
| longeRNA-pp_change.txt | Differential expression results of promoter-proximal regions of long eRNAs across two conditions |
| longeRNA-gb_change.txt | Differential expression results of gene body regions of long eRNAs across two conditions |
| longeRNA-pindex_change.txt | Differential expression results of pausing index of long eRNAs across two conditions |
| signal_around_enhancer-center.pdf | PROseq signal around enhancer center for all samples |
Usage
You can add the eNRSA package folder to your $PATH, allowing you to run the scripts by their names without specifying the full path. e.g. suppose your eNRSA package is under
/home/user1/eNRSA
export PATH=$PATH:/home/user1/eNRSA
Example 1 - nascent transcriptional profile for known genes
pause_PROseq.py -in1 INF_EV_0hr_1-R1.bam INF_EV_0hr_2-R1.bam -in2 INF_Cre_0hr_1-R1.bam INF_Cre_0hr_2-R1.bam -output /nobackup/test-eNRSA/ -org hg38
The parameters are explained as follows.
Parameters |
Meaning |
|---|---|
pause_PROseq.py |
The command to profile nascent transcritional profile for known genes. |
-in1 INF_EV_0hr_1-R1.bam INF_EV_0hr_2-R1.bam |
The alignment files for condition one - control. |
-in2 INF_Cre_0hr_1-R1.bam INF_Cre_0hr_2-R1.bam |
The alignment files for condition two - case. |
-output /nobackup/test-eNRSA/ |
The directory for outputs. |
-org hg38 |
The organism. |
For all available parameters, see the Options.
Example 2 - nascent transcriptional profile for known genes with samples from different batches
pause_PROseq.py -in1 INF_EV_0hr_1-R1.bam INF_EV_0hr_2-R1.bam -in2 INF_Cre_0hr_1-R1.bam INF_Cre_0hr_2-R1.bam -output /nobackup/test-eNRSA/ -org hg38 -batch b1 b2 b1 b2
The parameters are explained as follows.
Parameters |
Meaning |
|---|---|
-batch b1 b2 b1 b2 |
The batch label for each sample. |
You can also specify the samples and batches using design table. For example,
pause_PROseq.py -design /nobackup/test-eNRSA/test-design.txt -output /nobackup/test-eNRSA/ -org hg38
Example 3 - nascent transcriptional profile for known genes using user-defined GTF file
pause_PROseq.py -in1 INF_EV_0hr_1-R1.bam INF_EV_0hr_2-R1.bam -in2 INF_Cre_0hr_1-R1.bam INF_Cre_0hr_2-R1.bam -output /nobackup/test-eNRSA/ -gtf /nobackup/test-eNRSA/test-gtf.txt
The parameters are explained as follows.
Parameters |
Meaning |
|---|---|
-gtf /nobackup/test-eNRSA/test-gtf.txt |
The path to GTF file. |
Example 4 - nascent transcriptional profile for enhancers
Note
eNRA.py depends on the results of pause_PROseq.py, please run pause_PROseq.py first.
eRNA.py -design /nobackup/test-eNRSA/test-design.txt -output /nobackup/test-eNRSA/ -org hg38
For all available parameters, see the Options
Container Usage
Docker
Add the
-vargument to docker command to mount the input and output disk on the host, e.g. you are working under /data directory, you need to add-v /data:/data, otherwise, your data won’t be recognized inside of dockerAbsolute path is required for all the files, including
-in1/-in2and-gtf/-fa(if applicable), the relative path doesn’t work, because docker uses isolated filesystem, and the working path inside and outside of docker are different.
docker run -v /data:/data chccode/enrsa:latest pause_PROseq.py <options>
docker run -v /data:/data chccode/enrsa:latest eRNA.py <options>
Singularity
Singularity typically mounts the current working directory automatically. However, if you encounter mounting issues, you can explicitly bind the input and output directories using the -B option.
singularity exec -B /data:/data enrsa.sif pause_PROseq.py <options>
singularity exec -B /data:/data enrsa.sif eRNA.py <options>
Options
pause_PROseq.py
| Parameters | Meaning |
|---|---|
| -in1 [bed/bam] | required, read alignment files in bed (6 columns) or bam format for condition1, each file is separated by space |
| -in2 [bed/bam] | read alignment files in bed (6 columns) or bam format for condition2, each file is separated by space (It is NOT required to have the same number of samples for each condition. The differential analysis is performed on condition 2 vs. 1.) |
| -design / -design_table [file] | desgin table in tsv format. 2 sections, first section is the sample information, 2 or 3 columns. For details, please refer to Design Table part |
| -batch / -b | Optional, if -in1 / -in2 is specified, and the need to adjust the batch effect, add the batch label for each sample. the total argument number should match with in1 + in2 |
| -gtf | user specified GTF file, if not specified, will use the default GTF file for the organism |
| -fa | Full path for the fasta file for the genome. If not specified, will search under the fa folder under the package directory. |
| -organism / -org | The genome build of the alignment files. Built-in = hg19, hg38, mm10, mm39, dm3, dm6, ce10, or danRer10. if other organism used, must also specify the -gtf and -fa |
| -pwout / -output [string] | required, output/work directory |
| -f1 [string] | normalization factors for samples of condition1, separated by space. If not specified, we will use DESeq2 default method to normalize |
| -f2 [string] | normalization factors for samples of condition2, same to -f1 |
| -up [int] | defines the upstream of TSS as promoter (bp, default: 500) |
| -down [int] | defines the downstream of TSS as promoter (bp, default: 500) |
| -gb [int] | defines the start of gene body density calculation (bp, default: 1000) |
| -min_gene_len [int] | defines the minimum length of a gene to perform the analysis (bp, default: 1000). Genes with length less than this will be ignored. |
| -window [int] | defines the window size (bp, default: 50) |
| -step [int] | defines the step size (bp, default: 5) |
| -sorted | Flag, the input bed files are sorted, skip the sorting step |
| -tts_down | TTS downstream length for detecting readthrough |
| -h | help message |
eRNA.py
| Parameters | Meaning |
|---|---|
| -in1 [bed/bam] | required, read alignment files in bed (6 columns) or bam format for condition1, each file is separated by space |
| -in2 [bed/bam] | read alignment files in bed (6 columns) or bam format for condition2, each file is separated by space (It is NOT required to have the same number of samples for each condition. The differential analysis is performed on condition 2 vs. 1.) |
| -design / -design_table [file] | desgin table in tsv format. 2 sections, first section is the sample information, 2 or 3 columns. For details, please refer to Design Table part |
| -gtf | user specified GTF file, if not specified, will use the default GTF file for the organism |
| -fa | Full path for the fasta file for the genome. If not specified, will search under the fa folder under the package directory. |
| -organism / -org | The genome build of the alignment files. Built-in = hg19, hg38, mm10, mm39, dm3, dm6, ce10, or danRer10. if other organism used, must also specify the -gtf and -fa |
| -pwout / -output [string] | required, output/work directory |
| -cutoff [int] | distance cutoff for divergent transcripts for enhancer detection (bp, default: 400) |
| -distance [int] | distance within which two eRNAs are merged (bp, default: 500) |
| -le [int] | length cutoff for long-eRNA identification (bp, default: 10000) |
| -filter [0,1] | whether to filter for enhancers: 0 (not filter) or 1 (filter), deflault: 1 |
| -dtss [int] | if filter enhancers, the minimum distance from enhancer to TSS (Transcription Start Site) (bp, default: 2000) |
| -dtts [int] | if filter enhancers, the minimum distance from enhancer to TTS(Transcription Termination Site) (bp, default: 20000) |
| -wd [int] | distance within which associate active genes of enhancer (bp, default: 50000) |
| -pri | flag, if set, will prioritize enhancer. default is false |
| -peak [file] | required if -pri set. ChIP-seq peak file for the regulator of interest in bed format |
| -lk ["pp", "gb", "pindex"] | valid when -pri is set for two conditions. Transcriptional changes to be used for enhancer prioritization, the value can be pp (changes in promoter-proximal region), gb (changes in gene body region), or pindex (changes in pausing index) (default: gb) |
| -direction [-1, 0, 1] | valid when -pri set for two conditions. The expected simultaneous change of expression between enhancers and target genes. 1: the expression changes of enhancers and target genes are in the same direction; -1: the expression changes of enhancers and target genes are in the opposite direction; 0: the expression changes of enhancers and target genes are either in the same or in the opposite direction (default: 0) |
| -wt/-weight [float] | valid when -pri set for two conditions. Weight to balance the impact of binding and function evidence. The higher the weight, the bigger impact does the binding evidence have (default: 0.5, i.e., binding and functional evidence have equal impact on enhancer prioritization) |
| -fdr [float] | valid when -pri set for two conditions. Cutoff to select significant transcriptional changes (default: FDR < 0.05). Use Foldchange instead if no FDR is generated (default: Foldchange > 1.5) |
| -sorted | Flag, the input bed files are sorted, skip the sorting step |