Our Services
VANGARD provides standard analysis for RNA-Seq, DNA-Seq, ChIP-Seq, and GRO/PRO-Seq, which implements "best practices" pipeline for the sequencing data provided.
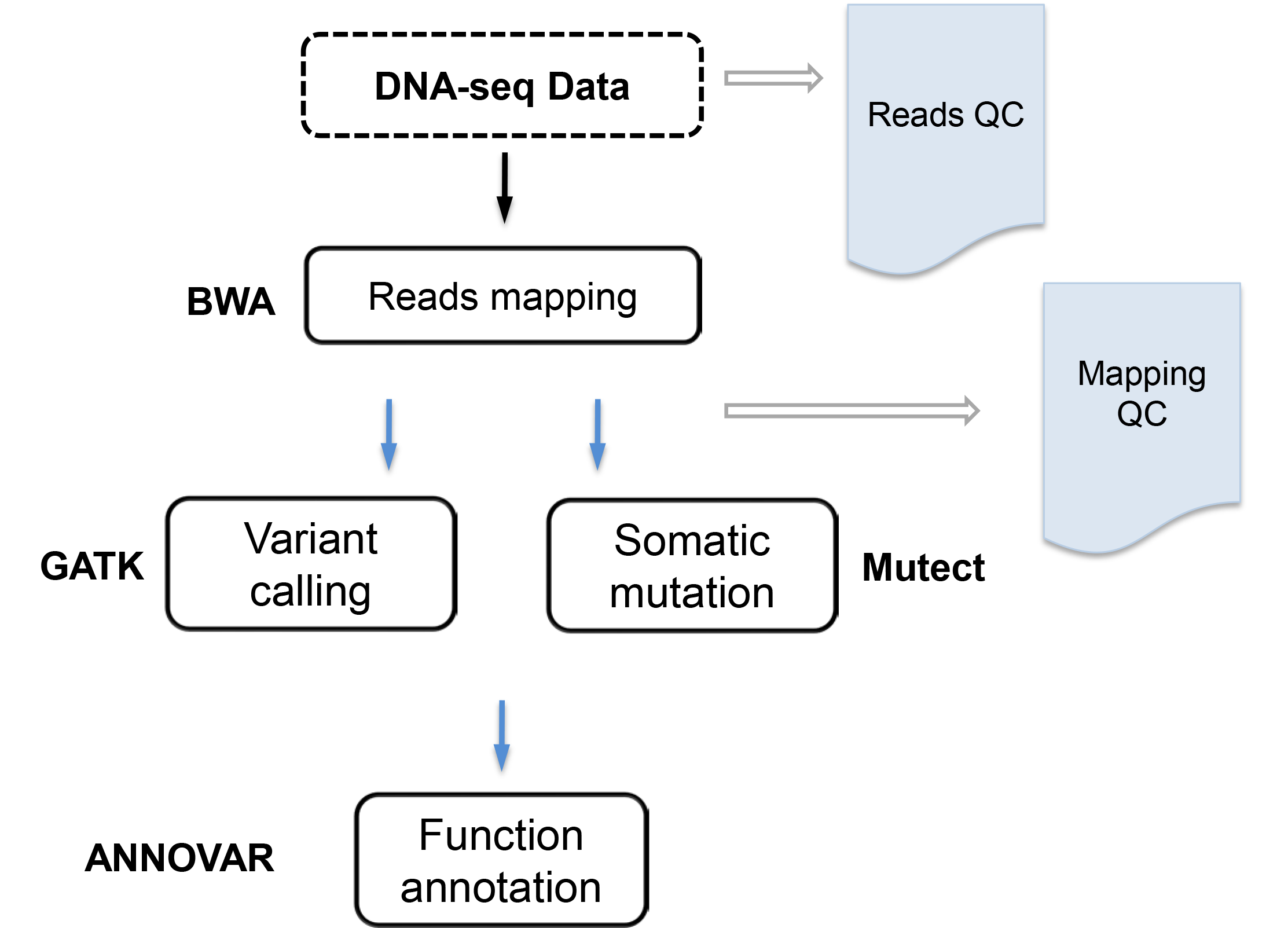
DNA-Seq Analysis
The goal is to call variants or somatic mutations. DNA-seq data are mapped to the reference genome using BWA1, variants are called using GATK2 Best practice, somatic mutation are identified by Mutect3, and variants/somatic mutations are functionally annotated by ANNOVAR4. VANGARD will perform quality check, including raw reads quality (reads QC), mapping quality (mapping QC) to ensure high quality results.
References
1. Li, H. and Durbin, R. (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics, 26, 589-595.
2. McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly, M. et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res, 20, 1297-1303.
3. Cibulskis, K., Lawrence, M.S., Carter, S.L., Sivachenko, A., Jaffe, D., Sougnez, C., Gabriel, S., Meyerson, M., Lander, E.S. and Getz, G. (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol, 31, 213-219.
4. Wang, K., Li, M. and Hakonarson, H. (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res, 38, e164.