Our Services
VANGARD provides standard analysis for RNA-Seq, DNA-Seq, ChIP-Seq, and GRO/PRO-Seq, which implements "best practices" pipeline for the sequencing data provided.
RNA-Seq Analysis
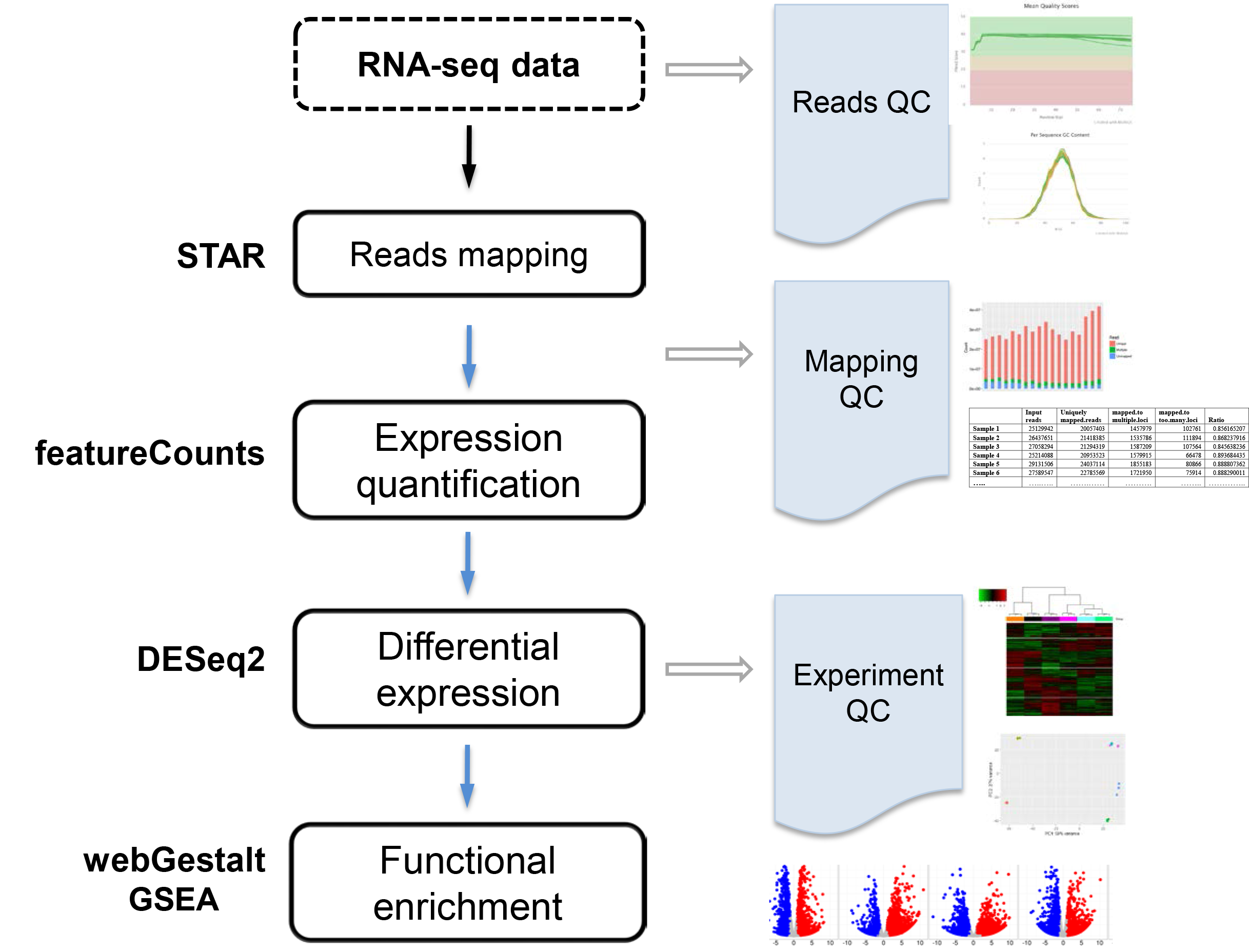
The goal is to identify differentially expressed genes across conditions. RNA-seq data are mapped to the reference genome using STAR1, gene expression are quantified by featureCounts2, differentially expressed genes are identified by DESeq23, and enriched function or pathways are discovered by webGestalt4 and GSEA5. VANGARD will perform quality check, including raw reads quality (reads QC), mapping quality (mapping QC) and experiment quality (experiment QC) to ensure high quality results. VANGARD will summarize all the results in a report.
References
1. Dobin, A., Davis, C.A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15-21.
2. Liao, Y., Smyth, G.K. and Shi, W. (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923-930.
3. Love, M.I., Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 15, 550.
4. Wang, J., Vasaikar, S., Shi, Z., Greer, M. and Zhang, B. (2017) WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res.
5. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 102(43):15545-50.